DADA2: Fast and accurate sample inference from amplicon data with single-nucleotide resolution
The DADA2 1.26 release is live, with native support for ARM architectures such as the Apple M1/M2 chips! Release notes.
Installation
Binaries for the current release version of DADA2 (1.26) are available from Bioconductor. Note that you must have R 4.2.0 or newer, and Bioconductor version 3.16, to install the most current release from Bioconductor.
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("dada2", version = "3.16")If you wish to install the latest and greatest development version, or to install to earlier versions of R, see our from-source installation instructions.
Tutorials
Start here: The DADA2 tutorial goes through a typical workflow for paired end Illumina Miseq data: raw amplicon sequencing data is processed into the table of exact amplicon sequence variants (ASVs) present in each sample.
The DADA2 Workflow on Big Data goes through workflow optimized to run on large datasets (10s of millions to billions of reads).
An ITS-specific version of the DADA2 workflow identifies and verifiably removes primers on both ends of each ITS read, a key step due to the variable length of the ITS region.
Short demonstrations of assigning taxonomy and assigning species to sequences.
Benchmarking
Our manuscript introducing DADA2 (OA link) compares the accuracy of DADA2 and other methods on several mock community datasets.
We describe the broad advantages of exact sequence variants over OTUs in our recent open-access ISMEJ paper.
We evaluate the high accuracy and resolution that is achievable by sequencing the full-length 16S rRNA gene with PacBio and DADA2.
Further benchmarking of DADA2 against the methods evaluated in a recent QIIME1 benchmarking paper is available.
And sometimes a picture says a thousand word:
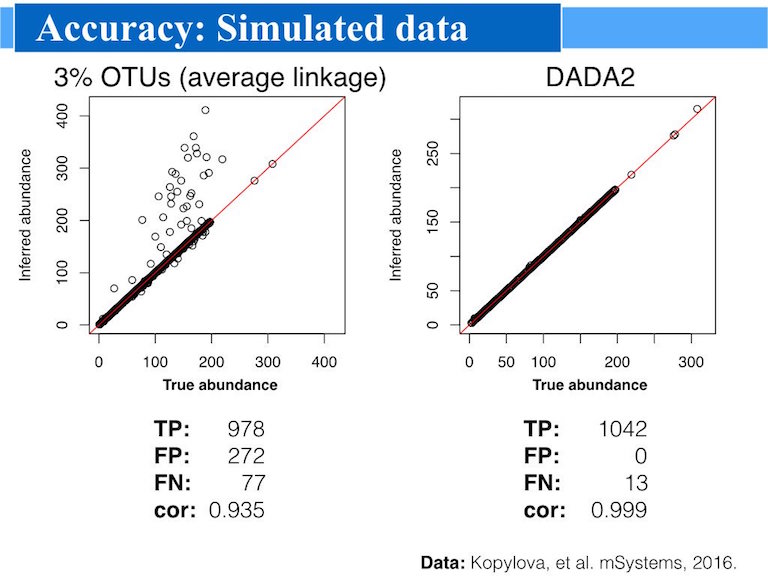
Advantages
Resolution: DADA2 infers exact amplicon sequence variants (ASVs) from amplicon data, resolving biological differences of even 1 or 2 nucleotides.
Accuracy: DADA2 reports fewer false positive sequence variants than other methods report false OTUs.
Comparability: The ASVs output by DADA2 can be directly compared between studies, without the need to reprocess the pooled data.
Computational Scaling: The compute time of DADA2 scales linearly sample number, and memory requirements are essentially flat.
Open Source: DADA2 is licensed under the LGPL version 3.
Support and Development
Planned feature improvements are publicly catalogued at the main DADA2 development site on github; specifically on the issues tracker for DADA2. If the feature you are hoping for is not listed, you are welcome to add it as a feature request on this page.
Bug reports and problems using DADA2 are also welcome on the issues tracker. We prefer posting to the issue tracker over email as these posts are searchable by other users who may experience the same problems.
How?
Accuracy: DADA2’s crucial advantage is that it uses more of the data. The DADA2 error model incorporates quality information, which is ignored by all other methods after filtering. The DADA2 error model incorporates quantitative abundances, whereas most other methods use abundance ranks if they use abundance at all. The DADA2 error model identifies the differences between sequences, eg. A->C, whereas other methods merely count the mismatches. DADA2 can parameterize its error model from the data itself, rather than relying on previous datasets that may or may not reflect the PCR and sequencing protocols used in your study.
Performance: DADA2’s computational scaling gains come from the fact that it infers sequences exactly rather than constructing OTUs. De novo OTUs cannot be compared across samples unless all samples were pooled during OTU construction. However, exact sequences are comparable across samples, as exact sequences are consistent labels. Thus DADA2 can analyze each sample independently, resulting in linear scaling with sample number and trivial parallelization.
Maintained by Benjamin Callahan (benjamin DOT j DOT callahan AT gmail DOT com)
Documentation License: CC-BY 4.0